一个革命性的免费软件平台,助力您的计算化学研究。 PsiStack通过将各种强大的软件集成到一个用户友好的平台上,彻底改变了计算化学软件的管理。让复杂且耗时的安装和部署过程现在变得轻而易举。 PsiStack支持Amber, Gromacs, OpenMM, PsiCode, XTB, Plumed, PySCF等计算化学软件在多种操作系统上的快速安装和部署,免去逐一编译安装的烦恼,并简化软件更新和维护。 在线咨询 将您的研究提升到新的高度 在计算化学中管理各种软件包及其复杂的相互依赖关系(如GCC和FFTW)是一件极其繁琐的工作。此外,跟踪科学软件的更新(这些软件通常有特定的版本要求)也非常耗费精力,并可能分散您的核心研究目标。PsiStack通过为您处理软件依赖性和更新,减轻了这些负担。PsiStack兼容运行CentOS、Rocky、Ubuntu及类似操作系统的工作站、服务器和集群。目前,它支持广泛的流行工具,包括Amber、Gromacs、NAMD、OpenMM、LAMMPS、PLUMED、Autodock、PSI4、xTB、PySCF、CP2K、Quantum Espresso等。SP团队致力于不断扩展PsiStack的功能,引入更多的软件包和支持,让您可以专注于真正重要的事情——您的研究。 PsiStack通过以下优势,帮助计算化学家专注于研究,轻松驾驭创新的未来 一站式工具平台整合从量子力学到分子动力学的多样化开源软件,包括流行的工具和框架,所有都在 PsiStack仓库中 迅捷的安装部署以最少的设置快速配置您的工作站、服务器或集群,无缝加载PsiStack存储库 持续自动更新软件定期引入领域内新软件并确保现有软件更新至最新版本,降低运维成本 安全可靠的数据传输通过单向HTTP连接规避防火墙限制,确保本地数据存储的安全性 高效的按需加载通过按需加载优化本地资源使用,满足精确需求 动态多版本支持 适应同一软件的多版本,满足当前版本和旧版本不同需求 如何安装 通过在终端中运行以下内容来设置 PsiStack 的客户端:$ curl -s https://stack.singleparticle.cloud/PsiStack-install.sh | sudo sh安装后,请先登出,然后重新登录您的计算机。运行命令列出所有可用软件。 有关安装过程的更多详细信息,您可以参考视频指南。 软件使用说明 列出所有软件$ module availOpenMM$ module load OpenMM/8.0.0-foss-2022a-CUDA-11.8.0PySCF$ module load PySCF/2.1.1-foss-2022aPSI4$ module load PSI4/1.7-foss-2022aAmberTools$ module load AmberTools/22.3-foss-2022a GROMACS$ module load GROMACS/2023.1-foss-2022a-CUDA-11.8.0PLUMED$ module load PLUMED/2.8.1-foss-2022a
 一个革命性的免费软件平台,助力您的计算化学研究。
一个革命性的免费软件平台,助力您的计算化学研究。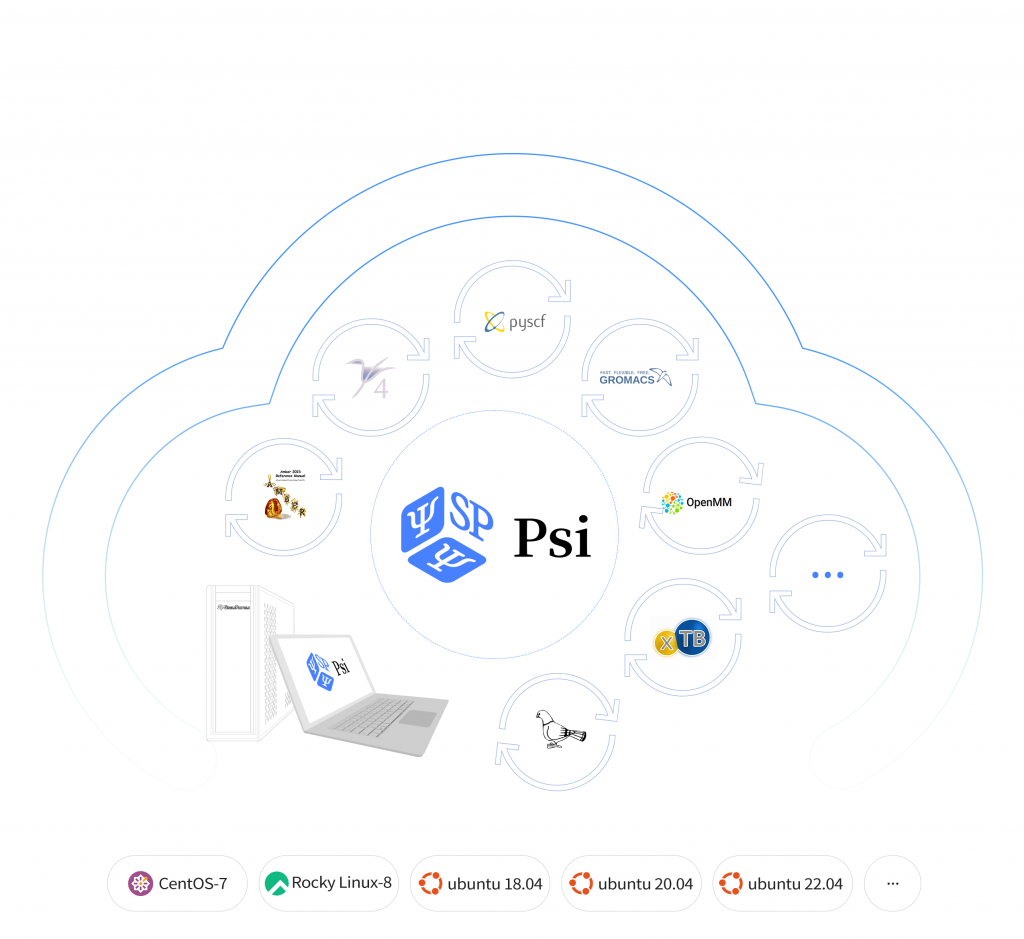 一个革命性的免费软件平台,助力您的计算化学研究。
一个革命性的免费软件平台,助力您的计算化学研究。